Best Methods for Predicting Hybridization Tm
Myth 2: Best Methods for Predicting Hybridization Tm
Best methods for predicting hybridization Tm are essentially equivalent in accuracy. The melting temperature, Tm, of duplex formation is usually defined as the temperature at which half the available strands are in the double-stranded state (or folded state for unimolecular transitions) and half the strands are in the “random coil” state. We will see later (i.e., myth 3) that this definition is not general and that the Tm itself is not particularly useful for PCR design. Over the past 45 years (1960–2005), there have been a large number of alternative methods for predicting DNA duplex Tm that have been published. The simplest equation based on base content is the “Wallace rule” (19):
![]()
This equation neglects many important factors: Tm is dependent on strand concentration, salt concentration, and base sequence. Typical error for this simple method compared with experimental Tm is greater than 15C, and thus this equation is not recommended. A somewhat more advanced base content model is given by (20,21):
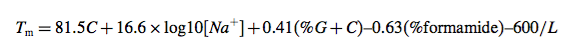
where L is the length of the hybrid duplex in base pairs. Maxim Frank- Kamenetskii provided a more accurate polymer salt dependence correction in 1971 (22). Nonetheless, Eq. 30 was derived for polymers, which do not include bimolecular initiation that is present in oligonucleotides, does not account for sequence dependent effects, and does not account for terminal end effects that are present in oligonucleotide duplexes (7). Thus, this equation works well for DNA polymers, where sequence-dependent effects are averaged out, and long duplexes (greater than 40 base pairs) but breaks down for short oligonucleotide duplexes that are typically used for PCR. Both these simple equations are inappropriate for PCR design. Further work suggested (wrongly) that the presence of mismatches in DNA polymers can be accounted for by decreasing the Tm by 1C for every 1% of mismatch present in the sequence (20,23). Based on a comprehensive set of measurements from my laboratory (9), we now know that this is highly inaccurate (mismatch stability is very sequence dependent), and yet some commercial packages continue to use it.
The appropriate method for predicting oligonucleotide thermodynamics is the NN model (2,7). The NN model is capable of accounting for sequence- dependent stacking as well as bimolecular initiation. As of 1996, there were at least eight sets of NN parameters of DNA duplex formation in the literature, and it was not until 1998 that the different parameter sets were critically evaluated and a “unified NN set” was developed (7). Several groups (7,24) came to the same conclusion that the 1986 parameters (25) are unreliable. Unfortunately, the wrong 1986 parameters are still present in some of the most widely used packages for PCR design (namely, Primer3, OLIGO, and VectorNTI). Table 1 compares the quality of predictions for different parameter sets.
The results in Table 1 clearly demonstrate that the 1986 NN set is unreliable and that the PCR community should abandon their use. The fact that many scientists have used these inaccurate parameters to design successful PCRs is a testament to the robustness of single-target PCR and the availability of optimization of the annealing temperature in PCR to improve amplification efficiency despite wrong predictions (see myth 1). However, as soon as one tries to use the old parameters to design more complicated assays such as multiplex PCR, real-time PCR, and parallel PCRs, then it is observed that the old parameters fail badly. The use of the “unified NN parameters,” on the contrary, results in much better PCR designs with more predictable annealing behavior and thereby enables high-throughput PCR applications and also multiplex PCR.
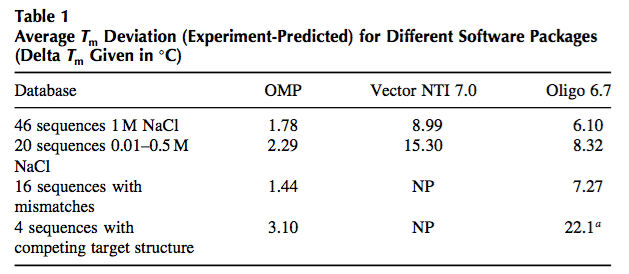
NP, calculation not possible with Vector NTI. a Oligo cannot predict target folding so the number given is for the hybridization neglecting target folding.
Furthermore, the unified NN parameters were extended by my laboratory to allow for accurate calculation of mismatches, dangling ends, salt effects, and other secondary structural elements, all of which are important in PCR (2).