Why BLAST Search is a Myth for Determining the Specificity of a Primer
Myth 5: A BLAST Search Is the Best Method for Determining the Specificity of a Primer
To minimize mispriming, several PCR texts suggest performing a BLAST search, and such capability is a part of some primer design packages such as GCG and Vector NTI and Visual-OMP. However, a BLAST search is not the appropriate screen for mispriming because sequence identity is not a good approximation to duplex thermodynamics, which is the proper quantity that controls primer binding. For example, BLAST scores a GC and an AT pair identically (as matches), whereas it is well known that base pairing in fact depends on both the G+C content and the sequence, which is why the NN model is most appropriate. In addition, different mismatches contribute differently to duplex stability.
For example, a G−G mismatch contributes as much as −22kcal/mol to duplex stability at 37C, whereas a C−C mismatch can destabilize a duplex by as much as +25 kcal/mol. Thus, mismatches can contribute G over a range of 4.7 kcal/mol, which corresponds to factor of 2000 in equilibrium constant.
In addition, the thermodynamics of DNA–DNA duplex formation are quite different than that of DNA–RNA hybridization. Clearly, thermodynamic parameters will provide better prediction of mispriming than sequence similarity. BLAST also uses a minimum 8nt “word length,” which must be a perfect match; this is used to make the BLAST algorithm fast, but it also means that BLAST will miss structures that have fewer than eight consecutive matches. As GT, GG, and GA mismatches are stable and occur commonly when a primer is scanned against an entire genome, such a short word length can result in BLAST missing thermodynamically important hybridization events.
BLAST also does not properly score the gaps that result in bulges in the duplexes. DNA Software, Inc. is developing a new algorithm called ThermoBLAST that retains the computational efficiency of BLAST so that searches genomic can be accomplished rapidly but uses thermody- namic scoring for base pairs, dangling end, single mismatches, bulges, tandem mismatches, and other motifs. Figure 10 gives some examples of strong hybridization that would be missed by BLAST but detected by ThermoBLAST. The computational efficiency of ThermoBLAST is accomplished using a variant of the bimolecular dynamic programming algorithm that was invented at DNA Software, Inc.
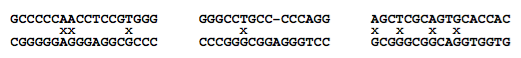
Fig. 10. Three hybridized structures that BLAST misses due to the word length limit of eight. All the structures shown are thermodynamically stable under typical PCR buffer conditions. Note the mismatches (denoted by “x”) and bulges (denoted by a gap in the alignment).