Author: joseph@dnasoftware.com
Multiplex PCR Optimization
Myth 7: Multiplex PCR Can Succeed by Optimization of Individual PCRs
Not too many people believe this myth, and yet their actions are somewhat irrational as they proceed to immediately use that approach to try to experimentally optimize a multiplex PCR. It is true that well-designed single-target PCRs are useful for developing a multiplex reaction, but for a variety of reasons, this approach alone is too simplistic. The most common experimental approach to optimizing a multiplex PCR design is shown in Fig. 11, as suggested by Henegariu et al. (30). This is a laborious procedure that has a high incidence of failure even after extensive experimentation. The core of this approach is to first optimize the single-target amplifications and then to iteratively combine primer sets to determine which primer sets are incompatible and also to try to adjust the thermocycling or buffer conditions. With such an approach, the optimization of a 10-plex PCR typically takes a PhD level scientist 3–6 months (or more), with a significant chance of failure anyway.
Why does the experimental approach fail? The answer is that there are simply too many variables (i.e., many different candidate primers and targets) in the system and that the variables interact with each other in non-intuitive ways. Anyone who has actually gone through this experimental exercise will attest to the exasperation and disappointment that occurs when 7 of 10 of the amplicons are being made efficiently (after much work) only to have some of them mysteriously fail when an eighth set of primers is added. The approach of trying to adjust the thermocycling or buffer conditions is also doomed to failure because the changes affect all the components of the system in

Fig. 11. Multiplex PCR optimization guidelines suggested by Henegariu et. al. (30).
different ways. For example, increasing the annealing temperature might be a fine way to minimize primer dimer artifacts (which can be a big problem in multiplex PCR), but then some of the weaker primers start to bind inefficiently. Subsequent redesign of those weak primers might then make them interact with another component of the reaction to form mishybridized products or new primer dimers, or cause that amplicon to take over the multiplex reaction.
The mystery could have been prevented (or at least minimized) with the use of a proper software tool. First, proper design if the single-target PCRs leads to improved success when used in multiplex. Second, software can try millions of combinations with much more complete models of the individual components (as described throughout this chapter) and use more complete modeling of the interactions within the whole system, whereas a human can only try a few variables before getting exhausted.
Amplification Efficiency for primers Is Not Exponential
Myth 6: At the End of PCR, Amplification Efficiency Is Not Exponential Because the Primers or NTPs Are Exhausted or the Polymerase Looses Activity
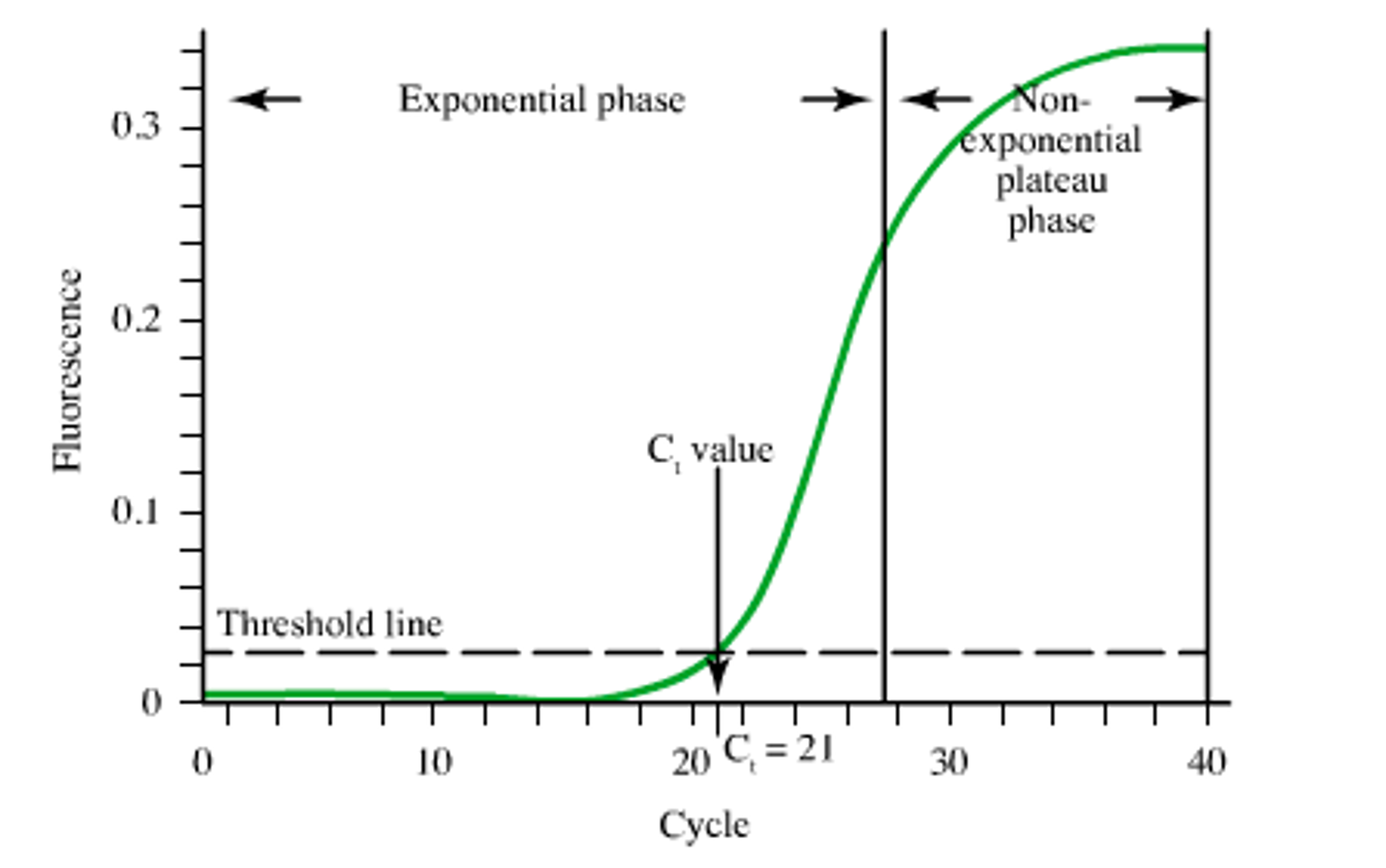 PCR amplification occurs with a characteristic “S” shape. During the early cycles of PCR, the amplification is exponential. During the later stages of PCR, saturation behavior is observed, and the amplification efficiency of PCR decreases with each successive cycle. What is the physical origin of the saturation and why is the explanation important for PCR design? Most practitioners of PCR believe that saturation is observed because either the primers or the NTPs are exhausted or the polymerase looses activity.
PCR amplification occurs with a characteristic “S” shape. During the early cycles of PCR, the amplification is exponential. During the later stages of PCR, saturation behavior is observed, and the amplification efficiency of PCR decreases with each successive cycle. What is the physical origin of the saturation and why is the explanation important for PCR design? Most practitioners of PCR believe that saturation is observed because either the primers or the NTPs are exhausted or the polymerase looses activity.
The idea of lost polymerase activity is historical. In the early days of PCR, polymerase enzymes did loose activity with numerous cycles of PCR. Modern thermostable engineered polymerases, however, are quite robust and exhibit nearly full activity at the end of a typical PCR. The idea of one or more of the NTPs or primers being limiting reagents is perfectly logical and consistent with chemical principles but is not correct for the concentrations that are usually used in PCR. Chemical analysis of the PCR mixture reveals that at the end of PCR there is usually plenty of primers and NTPs so that PCR should continue for further cycles before saturation is observed due to consumption of a limiting reagent. Experimentally, if you double the concentration of the primers, you do not observe twice the PCR product. Thus, the amplicon yield of PCR is usually less than predicted based on the primer concentrations.
What is causing the PCR to saturate prematurely? The answer is that double-stranded DNA is an excellent inhibitor of DNA polymerase. This can be demonstrated experimentally by adding a large quantity of non-extensible “decoy” duplex DNA to a PCR and comparing the result to a PCR without the added duplex. The result clearly shows that the reaction with added duplex DNA shows little or no amplification while the control amplifies normally. The reason why duplex DNA inhibits DNA polymerase is that the polymerase binds to the duplex rather than binding to the small quantity of duplex arising from the primers binding to target strands during the early cycles of PCR.
Application of the Inhibition Principle to Multiplex PCR Design
The concept of amplicon inhibition of PCR is particularly important for multiplex PCR design. Consider a multiplex reaction in which there are plenty of NTPs available. It is expected that if one of the amplicons is produced more efficiently than the others, then it will reach saturation and inhibit the polymerase from subsequently amplifying the other amplicons.
To achieve uniform amplification of the different targets, the primers must be designed to bind with equal efficiency to their respective targets. Binding equally does not mean “matched Tm’s.” This requires the use of accurate thermodynamic parameters (i.e., by not using the older methods for Tm prediction) and also accounting for the effects of competing equilibria, which requires the use of the coupled multi-state equilibrium model described in Subheadings 2.1 and 2.4 as well as the other principles described in this chapter.
Why BLAST Search is a Myth for Determining the Specificity of a Primer
Myth 5: A BLAST Search Is the Best Method for Determining the Specificity of a Primer
To minimize mispriming, several PCR texts suggest performing a BLAST search, and such capability is a part of some primer design packages such as GCG and Vector NTI and Visual-OMP. However, a BLAST search is not the appropriate screen for mispriming because sequence identity is not a good approximation to duplex thermodynamics, which is the proper quantity that controls primer binding. For example, BLAST scores a GC and an AT pair identically (as matches), whereas it is well known that base pairing in fact depends on both the G+C content and the sequence, which is why the NN model is most appropriate. In addition, different mismatches contribute differently to duplex stability.
For example, a G−G mismatch contributes as much as −22kcal/mol to duplex stability at 37C, whereas a C−C mismatch can destabilize a duplex by as much as +25 kcal/mol. Thus, mismatches can contribute G over a range of 4.7 kcal/mol, which corresponds to factor of 2000 in equilibrium constant.
In addition, the thermodynamics of DNA–DNA duplex formation are quite different than that of DNA–RNA hybridization. Clearly, thermodynamic parameters will provide better prediction of mispriming than sequence similarity. BLAST also uses a minimum 8nt “word length,” which must be a perfect match; this is used to make the BLAST algorithm fast, but it also means that BLAST will miss structures that have fewer than eight consecutive matches. As GT, GG, and GA mismatches are stable and occur commonly when a primer is scanned against an entire genome, such a short word length can result in BLAST missing thermodynamically important hybridization events.
BLAST also does not properly score the gaps that result in bulges in the duplexes. DNA Software, Inc. is developing a new algorithm called ThermoBLAST that retains the computational efficiency of BLAST so that searches genomic can be accomplished rapidly but uses thermody- namic scoring for base pairs, dangling end, single mismatches, bulges, tandem mismatches, and other motifs. Figure 10 gives some examples of strong hybridization that would be missed by BLAST but detected by ThermoBLAST. The computational efficiency of ThermoBLAST is accomplished using a variant of the bimolecular dynamic programming algorithm that was invented at DNA Software, Inc.
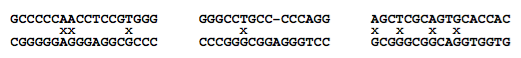
Fig. 10. Three hybridized structures that BLAST misses due to the word length limit of eight. All the structures shown are thermodynamically stable under typical PCR buffer conditions. Note the mismatches (denoted by “x”) and bulges (denoted by a gap in the alignment).
Primer Dimer Artifacts Are Due to Dimerization
Myth 4: “Primer Dimer” Artifacts Are Due to Dimerization of Primers
A common artifact in PCR is the amplification of “primer dimers.” The most common conception of the origin of primer dimers is that two primers hybridize at their 3′-ends (see Fig. 8). DNA polymerase can bind to such species and extend the primers in both directions to produce an undesired product with a length that is slightly less than the sum of the lengths of the forward and reverse primers. This mechanism of primer dimerization is certainly feasible and can be experimentally demonstrated by performing thermocycling in the absence of target DNA. This mechanism can also occur when the desired ampli- fication of the target is inefficient (e.g., when one of the primers is designed to bind to a region of the target that is folded into a stable secondary structure). Therefore, most PCR design software packages check candidate primers for 3′-complementarity and redesign one or both of them if the thermodynamic
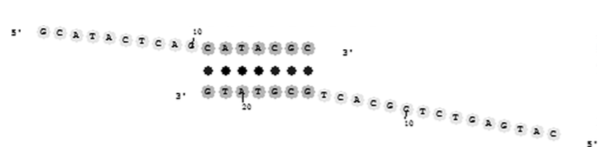
Fig. 8. Primer dimer hybridized duplex. Note that the 3′-ends of both primers are extensible by DNA polymerase.
stability of the hybrid is above some threshold. Another practical strategy to reduce primer dimer formation is to design the primer to have the last two nucleotides as AA or TT, which reduces the likelihood of a primer dimer structure with a stable hybridized 3′-end (29). For single-target PCR, two primers are present (FP and RP), and there are three different combinations of primer dimers that are possible FP–FP, RP–RP, and FP–RP. For multiplex PCR with N primers, there are NC2 pairwise combinations that are possible, and it becomes harder to redesign the primers so that all of them are mutually compatible. This becomes computationally challenging for large-scale multi- plexing. However, such computer optimization is only partially effective at removing the primer dimer artifacts in real PCRs. Why?
An Alternative Mechanism for Primer Dimer Artifacts
There are some additional observations that provide clues for an alternative mechanism for primer dimerization.
- Generally, homodimers (i.e., dimers involving the same strand) are rarely observed.
- Primer dimer artifacts typically occur at a large threshold cycle number (usually > 35 cycles), which is higher than the threshold cycle number for the desired amplicon.
- Primer dimers increase markedly when heterologous genomic DNA is added.
- Primer dimers are most often observed when one or both of the primers bind inefficiently to the target DNA (e.g., due to secondary structure of the target or weak thermodynamics).
- When the primer dimers are sequenced, there are often a few extra nucleotides of mysterious origin in the center of the dimer amplicon.
Observations 1 and 2 suggest that DNA polymerase does not efficiently bind to or extend primer duplexes with complementary 3′-ends. Observation 2 could also be interpreted as meaning that the concentration of the primer duplex is quite low compared with the normal primer-target duplex. In the early stages of PCR, however, observations 3 and 5 suggest that background genomic DNA may play a role in the mechanism of primer dimer formation. Observation 4 suggests that primer dimerization needs to occur in the early rounds of PCR to prevent the desired amplicon from taking over the reactions in the test tube. Figure 9 illustrates a mechanism that involves the genomic DNA in the early cycles of PCR and that provides an explanation for all five observations.
The mechanism presented in Fig. 9 can also be checked for by computer, but searching for such a site in a large genome can be quite computationally demanding. The ThermoBLAST algorithm developed by DNA Software, Inc. can meet this challenge (see myth 5).
Additional Concerns for Primer Dimers
Two primers can sometimes hybridize using the 5′ end or middle of the sequences. Such structures are not efficiently extensible by DNA polymerase. Such 5′-end primer hybrids, however, can in principle affect the overall equilibrium for hybridization, but generally, this is a negligible effect that is easily minimized by primer design software (i.e., if a primer is predicted to form a significant interaction with one of the other primers, then one or both of the primers are redesigned to bind to a shifted location on the target). If a polymerase is used that has exonuclease activity (e.g., Pfu polymerase), then it is possible that hybridized structures that would normally be non-extensible might be chewed back by the exonuclease and create an extensible structure. Indeed, it is observed that PCRs done with enzymes that have exonuclease

Fig. 9. Genomic DNA can participate in the creation of both the desired amplicon and the primer dimerization artifact. Notice that despite the presence of a few mismatches, denoted by “x,” the middle and 5′-ends of the primers are able to bind to the target stronger than they would bind to another primer molecule. Note that this mechanism does not require very strong 3′-end complementarity of the primers P1 and P2. Instead, this mechanism requires that sites for P1 and P2 are close to each other.
activity have a much higher incidence of primer dimer formation and mishybridization artifacts. Thus, for PCR, “proofreading” activity can actually be harmful.
Designing Forward and Reverse Primers to Have Matching Tm

Myth 3: Designing Forward and Reverse Primers to Have Matching Tm’s Is the Best Strategy to Optimize for PCR
Nearly all “experts” in PCR design would claim to believe in myth 3. Most current software packages base their design strategy on this myth. Some careful thought, however, quickly reveals the deficiencies of that approach. The Tm is the temperature at which half the primer strands are bound to target. This provides intuitive insight for very simple reactions, but it does not reveal the behavior (i.e., the amount of primer bound to target) at the annealing temperature. The PCR annealing temperature is typically chosen to be 10 C below the Tm . However, different primers have different H of binding, which results in different slopes at the Tm of the melting transition. Thus, the hybridization behavior at the Tm is not the same as the behavior at the annealing temperature. The quantity that is important for PCR design is the amount of primer bound to target at the annealing temperature. Obtaining equal primer binding requires that the solution of the equilibrium equations as discussed in Subheading 2.1. If the primers have an equal concentration of binding, then they will be equally extended by DNA polymerase, resulting in efficient amplification. This principle is illustrated in Fig. 5. The differences in primer binding are amplified with each cycle of PCR, thereby reducing the amplification efficiency and providing opportunity for artifacts to develop. The myth of designing forward and reverse primers with matched Tm’s is thus flawed. Nonetheless, as single-target PCR is fairly robust, such inaccuracies are somewhat tolerated, particularly if one allows for experimental optimization of the temperature cycling protocol for each PCR. In multiplex and other complex assays, however, the design flaws from matched Tm’s become crucial and lead to failure.
An additional problem with using two-state Tm’s for primer design is that they do not account for the rather typical case where target secondary structure competes with primer binding. Thus, the two-state approximation is typically invalid for PCR, and thus the two-state Tm is not directly related to the actual behavior in the PCR. The physical principle that does account for the effects of competing secondary structure, mishybridization, primer dimers, and so on is called “multi-state equilibrium,” as described in Subheading 2.4.
Below an alternative design strategy is suggested in which primers are carefully designed so that many PCRs can be made to work optimally at a single PCR condition, thereby enabling high-throughput PCR without the need

Fig. 5. Illustration of hybridization profiles of primers with two different design strategies. In the left panel, the Tm’s of primers are matched at 686C, but at the annealing temperature of 58C, primer B (squares) binds 87% and primer A (diamonds) binds 97%. This would lead to unequal hybridization and polymerase extension, thus reducing the efficiency of PCR. In the right panel, the G at 58C of the two primers is matched by redesigning primer B. The result is that both primers are now 97% bound, and thus optimal PCR efficiency would be observed. Notice that the Tm’s of the two primers are not equal in the right panel.
or temperature optimization. This robust strategy also lays the foundation for designing multiplex PCR with uniform amplification efficiency in which one must perform all the amplification reactions at the same temperature.
Application of the Multi-State Model to PCR Design
A typical single-stranded DNA target is not “random coil” nor do targets form a linear conformation as cartoons describing PCR often show (see Fig. 6). Instead, target DNA molecules (and also primers sometimes) form stable secondary structure (see Fig. 7). In the case of RNA targets, which are important for reverse-transcription PCR, the RNAs may be folded into secondary and tertiary structures that are much more stable than a typical random DNA sequence. If the primer is designed to bind to a region of the target DNA

Fig. 6. The two-state model for duplex hybridization. The single-stranded target and probe DNAs are assumed to be in the random coil conformation.

Fig. 7. The multi-state model for the coupled equilibrium involved in DNA hybridization. Most software only calculates the two-state thermodynamics (vertical transition). The competing target and primer structures, however, significantly affect the effective thermodynamics (diagonal transition). Note that G(effective) is not the simple sum of G(unfolding) and G(hybridization), but instead the sum must be weighted by the species concentrations, which can only be obtained by solving the coupled equilibria for the given total strand concentrations. Note that a more precise model would also include competing equilibria for primer dimerization and mismatch hybridization.
or RNA that is folded, then the folding must be broken before the primer can bind (see Fig. 7). This provides an energetic barrier that slows down the kinetics of hybridization and also makes the equilibrium less favorable toward binding. This can result in the complete failure of a PCR or hybridization assay (a false-negative test). Thus, it is desirable to design primers to bind to regions of the target that are relatively free of secondary structure. DNA secondary structure can be predicted using the DNA-MFOLD server or using Visual-OMP as described in our review (2). Simply looking at a DNA secondary structure does not always obviously reveal the best places to bind a primer. The reason why the hybridization is more complex than expected is revealed by some reasoning. Primer binding to a target can be thought to occur in three steps: (1) the target partially unfolds, (2) the primer binds, and (3) the remainder of the target rearranges its folding to accomplish a minimum energy state. The energy required to unfold structure in step 1 can sometimes be partially compensated by the structural rearrangement energy from step 3 (as shown in Fig. 7). Such rearrangement energy will help the equilibrium to be more favorable toward hybridization, but the kinetics of hybridization will still be slower than what would occur in a comparable open- target site. Note that the bimolecular structure shown in Fig. 7 shows the tails of the target folded. Visual-OMP allows the prediction of such structures, which is accomplished by a novel bimolecular dynamic programming algorithm.
To compute the equilibrium binding is also not obvious in multi-state reactions. We recommend solving the coupled equilibria for the concentrations of all the species. This is best done numerically as described in Subheading 2.4. The only PCR design software currently available that can solve the multi-state coupled equilibrium is Visual-OMP. Some recent work by Zuker (26) with partition functions is also applicable to the issue of multi-state equilibrium but to date has not been integrated into an automated PCR design software package. These considerations make the choice of the best target site non- obvious. Mismatch hybridization to an unstructured region can sometimes be more favorable than hybridization at a fully match site that is folded, thereby resulting in undesired false priming artifacts in PCR. Perhaps, it will come as a surprise to some that secondary structure in the primer is beneficial for specificity but harmful to binding kinetics and equilibrium. A practical way to overcome the complexity problem is to simply simulate the net binding charac- teristics of all oligos of a given length along the target—this is called “oligo walking.” Oligo walking is automatically done in the PCR design module of Visual-OMP and is also available in the RNA-STRUCTURE software from David Matthews and Douglas Turner (27,28).
Best Methods for Predicting Hybridization Tm
Myth 2: Best Methods for Predicting Hybridization Tm
Best methods for predicting hybridization Tm are essentially equivalent in accuracy. The melting temperature, Tm, of duplex formation is usually defined as the temperature at which half the available strands are in the double-stranded state (or folded state for unimolecular transitions) and half the strands are in the “random coil” state. We will see later (i.e., myth 3) that this definition is not general and that the Tm itself is not particularly useful for PCR design. Over the past 45 years (1960–2005), there have been a large number of alternative methods for predicting DNA duplex Tm that have been published. The simplest equation based on base content is the “Wallace rule” (19):
![]()
This equation neglects many important factors: Tm is dependent on strand concentration, salt concentration, and base sequence. Typical error for this simple method compared with experimental Tm is greater than 15C, and thus this equation is not recommended. A somewhat more advanced base content model is given by (20,21):
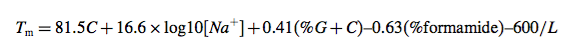
where L is the length of the hybrid duplex in base pairs. Maxim Frank- Kamenetskii provided a more accurate polymer salt dependence correction in 1971 (22). Nonetheless, Eq. 30 was derived for polymers, which do not include bimolecular initiation that is present in oligonucleotides, does not account for sequence dependent effects, and does not account for terminal end effects that are present in oligonucleotide duplexes (7). Thus, this equation works well for DNA polymers, where sequence-dependent effects are averaged out, and long duplexes (greater than 40 base pairs) but breaks down for short oligonucleotide duplexes that are typically used for PCR. Both these simple equations are inappropriate for PCR design. Further work suggested (wrongly) that the presence of mismatches in DNA polymers can be accounted for by decreasing the Tm by 1C for every 1% of mismatch present in the sequence (20,23). Based on a comprehensive set of measurements from my laboratory (9), we now know that this is highly inaccurate (mismatch stability is very sequence dependent), and yet some commercial packages continue to use it.
The appropriate method for predicting oligonucleotide thermodynamics is the NN model (2,7). The NN model is capable of accounting for sequence- dependent stacking as well as bimolecular initiation. As of 1996, there were at least eight sets of NN parameters of DNA duplex formation in the literature, and it was not until 1998 that the different parameter sets were critically evaluated and a “unified NN set” was developed (7). Several groups (7,24) came to the same conclusion that the 1986 parameters (25) are unreliable. Unfortunately, the wrong 1986 parameters are still present in some of the most widely used packages for PCR design (namely, Primer3, OLIGO, and VectorNTI). Table 1 compares the quality of predictions for different parameter sets.
The results in Table 1 clearly demonstrate that the 1986 NN set is unreliable and that the PCR community should abandon their use. The fact that many scientists have used these inaccurate parameters to design successful PCRs is a testament to the robustness of single-target PCR and the availability of optimization of the annealing temperature in PCR to improve amplification efficiency despite wrong predictions (see myth 1). However, as soon as one tries to use the old parameters to design more complicated assays such as multiplex PCR, real-time PCR, and parallel PCRs, then it is observed that the old parameters fail badly. The use of the “unified NN parameters,” on the contrary, results in much better PCR designs with more predictable annealing behavior and thereby enables high-throughput PCR applications and also multiplex PCR.
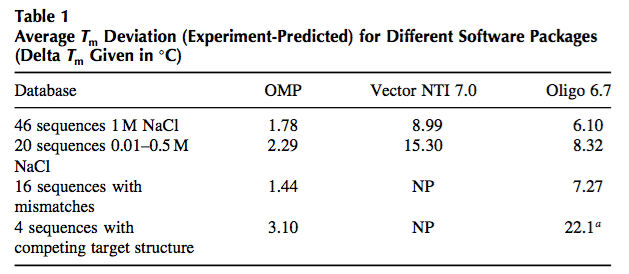
NP, calculation not possible with Vector NTI. a Oligo cannot predict target folding so the number given is for the hybridization neglecting target folding.
Furthermore, the unified NN parameters were extended by my laboratory to allow for accurate calculation of mismatches, dangling ends, salt effects, and other secondary structural elements, all of which are important in PCR (2).
PCR Working and Design Is Not Important
Myth 1: PCR Nearly Always Works and Design Is Not that Important
It might come as a surprise to many that despite the wide use and large investment, PCR in fact is still subject to many artifacts and environmental factors and is not as robust as would be desirable. Many of these artifacts can be avoided by careful oligonucleotide design. Over the last 10 years (1996–2006), I have informally polled scientists who are experts in PCR and asked: “What percentage of the time does a casually designed PCR reaction ‘work’ without any experimental optimization?” In this context, “work” means that the desired amplification product is made in good yield with a minimum of artifact products such as primer dimers, wrong amplicons, or inefficient amplification. By “casually designed,” I mean that typical software tools are used by an experienced molecular biologist. The consensus answer is 70–75%. If one allows for optimization of the annealing temperature in the thermocy- cling protocol (e.g., by using temperature gradient optimization), magnesium concentration optimization, and primer concentration optimization, then the consensus percentage increases to 90–95%. What is a user to do, however, in the 5–10% of cases where single-target PCR fails? Typically, they redesign the primers (without knowledge of what caused the original failure), resyn- thesize the oligonucleotides, and retest the PCR. Such a strategy works fine for laboratories that perform only a few PCRs. Once a particular PCR protocol is tested, it is usually quite reproducible, and this leads to the feeling that PCR is reliable. Even the 90–95% of single-target PCRs that “work” can be improved by using good design principles, which increases the sensitivity, decreases the background amplifications, and requires less experimental optimization. In a high-throughput industrial-scale environment, however, individual optimization of each PCR, redesigning failures, performing individualized thermocycling and buffer conditions, and tracking all these is a nightmare logistically and leads to non-uniform success. In multiplex PCR, all the targets are obviously amplified under the same solution and temperature cycling conditions, so there is no possibility of doing individual optimizations. Instead, it is desirable to have the capability to automatically design PCRs that work under a single general set of conditions without any optimization, which would enable parallel PCRs (e.g., in 384-well format) to be performed under the same buffer conditions and thermocycling protocol. Such robustness would further improve reliability of PCR in all applications but particularly in non-laboratory settings such as hospital clinics or field-testing applications.
Discovery of PCR
Shortly after the discovery of PCR, software for designing oligonucleotides was developed (12). Some examples of widely used primer design software (some of which are described in this book) include VectorNTI, OLIGO (12), Wisconsin GCG, Primer3 (13), PRIMO (14), PRIDE (15), PRIMERFINDER (http://arep.med.harvard.edu/PrimerFinder/PrimerFinderOverview.html), OSP (16), PRIMERMASTER (17), HybSIMULATOR (18), and PrimerPremiere. Many of these programs do incorporate novel features such as accounting for template quality (14) and providing primer predictions that are completely automated (14,15). Each software package has certain advantages and disadvantages, but all are not equal. They widely differ in their ease- of-use, computational efficiency, and underlying theoretical and conceptual framework. These differences result in varying PCR design quality. In addition, there are standalone Web servers that allow for individual parts of PCR to be predicted, notably DNA-MFOLD by Michael Zuker (http://www.bioinfo.rpi.edu/applications/mfold/old/dna/) and HYTHER by my laboratory (http://ozone3.chem.wayne.edu).
Why Is There a Need for Primer Design Software?
DNA hybridization experiments often require optimization because DNA hybridization does not strictly follow the Watson–Crick pairing rules. Instead, a DNA oligonucleotide can potentially pair with many sites on the genome with perhaps only one or a few mismatches, leading to false-positive results. In addition, the desired target sites of single-stranded genomic DNA or mRNA are often folded into stable secondary structures that must be unfolded to allow an oligonucleotide to bind. Sometimes, the target folding is so stable that very little probe DNA binds to the target, leading to a false-negative test. Various other artifacts include probe folding and probe dimerization. Thus, for DNA-based diagnostics to be successful, there is a need to fully understand the science underlying DNA folding and match versus mismatch hybridization. Achieving this goal has been a central activity of my academic laboratory as well as DNA Software, Inc.
Comparison of traditional quantitative pcr with computer quantitation
Comparison of traditional quantitative pcr with a computer-based quantitation algorithm for cmv from plasma specimens
Background
Difference between traditional quantitative pcr with computer-based quantitation algorithm for cmv. Traditional viral quantitation using cycle threshold (Ct) analysis is dependent on generating an accurate, valid calibration curve that is stable over time. The relationship between the Ct values obtained and the expected concentration for each calibrator is used to establish a formula to quantify viral concentrations in patient samples. Additionally, a fluorescence threshold is set to compare amplification cycles as an indirect measure of the starting DNA concentration. DNA Software, Inc. (Ann Arbor, MI) has developed a curve analysis algorithm (qPCR CopyCount) based on Poisson distribution that analyzes qPCR data to obtain an absolute DNA copy number without the need for a standard curve or use of Ct values. Our objectives were to compare the performance of qPCR CopyCount to traditional qPCR (Ct) for the quantitation of CMV from plasma and evaluate its feasibility for daily clinical use in a CMV plasma quantitation assay.
Methods
Fluorescence data from archived runs of our CMV assay, using Abbott ASR reagents on the Abbott m2000 system, were analyzed using CopyCount algorithm. CopyCount quantification was compared to original data using a calibration curve and Ct values. Nucleic acid quantitative standards (Qiagen), calibrators traceable to international units (CMVtc panel, Acrometrix), and patients were compared for linearity, precision, and accuracy.
Results
CopyCount results had a linear relationship with nucleic acid quantitative standard assigned values (r2>0.99; bias of log10 0.2 copies per reaction) and patient results by Ct method (r2>0.99). Raw CopyCount values do not account for extraction efficiency or concentration of the eluate, introducing a systematic bias between methods (bias -1.2 copies/ml). Evaluation of the CopyCount values from the extracted CMVtc panel demonstrated a linear relationship (r2>0.98) and provided a correction factor for extraction efficiency. Using this correction, the relationship between CopyCount and patient values remained linear (r2>0.99) with a reduced bias of log10 0.08 IU/ml. Precision of quantitative standard results (4 standards in duplicate over 10 runs) was a total standard deviation range of 0.05-0.15 log10 copies/rxn for CopyCount vs 0.02-0.16 log10 copies/ml for the current Ct method.
Conclusions
The CopyCount method performed similarly to the conventional Ct method for quantitation of CMV from patient samples and quantitative standards with excellent linearity and comparable precision. A correction factor or formula is required to DNA concentration in the PCR reaction to the concentration in the original specimen. A one-time calibration, using 1.5 copies per reaction as recommended by the software company to optimize the curve analysis formula, may further improve the accuracy of quantitation.
DNA Software releases ThermoBLAST Cloud Edition
DNA Software introduced the full commercial release of ThermoBLAST Cloud Edition (TB-CE). TB-CE provides a new standard for evaluating the target specificity of oligonucleotides.
DNAS gives webinar on qPCR CopyCount
DNAS gives webinar on qPCR CopyCount, click here to view video.